PhD Work
Reconstruction and Clustering with Graph optimization and Priors on Gene networks and Images
- Supervisors: Jean-Christophe Pesquet, Laurent Duval, Camille Couprie and Frédérique Bidard-Michelot
- Defense: July 3th, 2017 at 10h30 at ESIEE Paris (2 boulevard Blaise Pascal, Cité Descartes, BP 99, 93162 Noisy-le-Grand Cedex)
- Examining board: Pascal Frossard, Jean-Philippe Vert, Hugues Talbot, Stéphane Robin
Publications
- Aurélie Pirayre, Camille Couprie, Laurent Duval and Jean-Christophe Pesquet
- "BRANE Clust: Cluster-Assisted Gene Regulatory Network Inference Refinement"
- In IEEE/ACM Transactions on Computational Biology and Bioinformatics , March 2017
- [ DOI | Preprint | biorxiv | HAL | RNA-seq blog | OMIC tools | Software ]
- Aurélie Pirayre, Camille Couprie, Frédérique Bidard, Laurent Duval and Jean-Christophe Pesquet
- Dante Poggi-Parodi, Frédérique Bidard, Aurélie Pirayre, Thomas Portnoy, Corinne Blugeon, Bernhard Seiboth, Christian P. Kubicek, Stéphane Le Crom and Antoine Margeot
- "Kinetic transcriptome reveals an essentially intact induction system in a cellulase hyper-producer Trichoderma reesei strain"
- In Biotechnology for Biofuels , 2014, 7:173
- [ DOI ]
Abstract
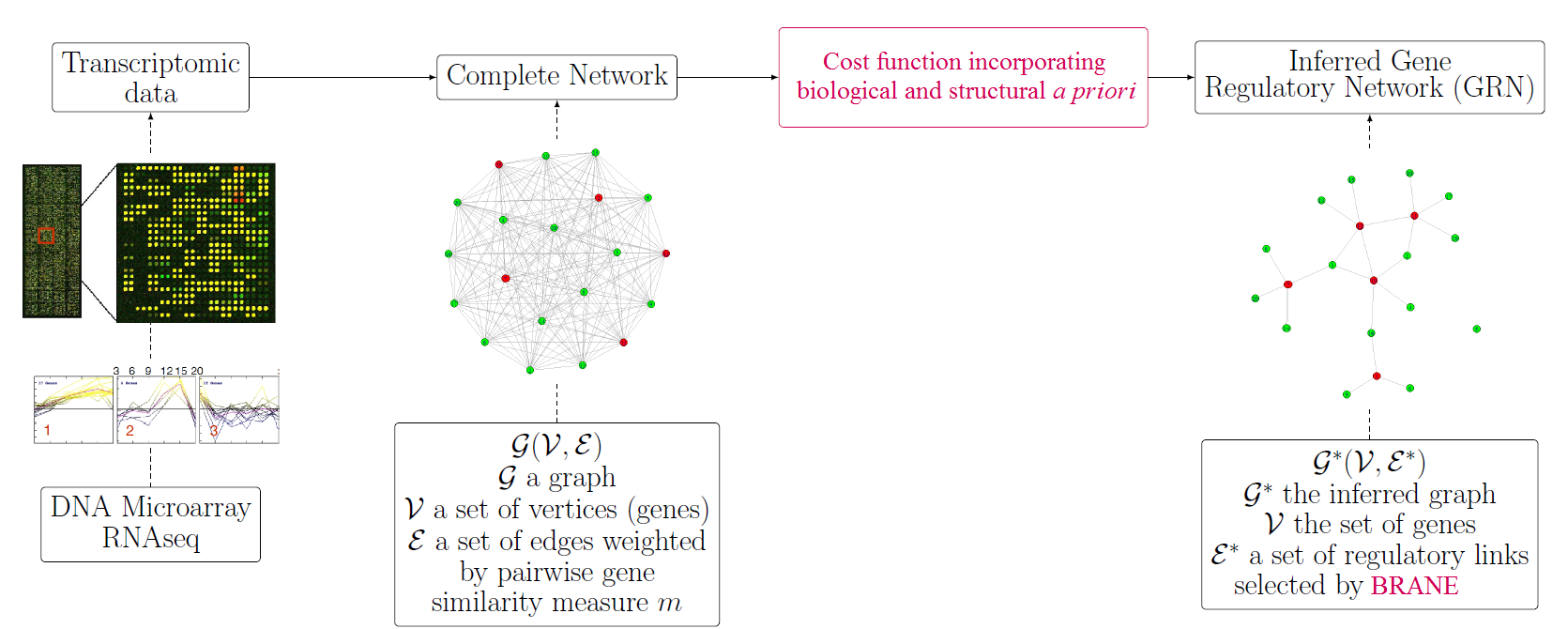
The discovery of novel gene regulatory processes improves the understanding of cell phenotypic responses to external stimuli for many biological applications, such as medicine, environment or biotechnologies. To this purpose, transcriptomic data are generated and analyzed from DNA microarrays or more recently RNAseq experiments. They consist in genetic expression level sequences obtained for all genes of a studied organism placed in different living conditions. From these data, gene regulation mechanisms can be recovered by revealing topological links encoded in graphs. In regulatory graphs, nodes correspond to genes. A link between two nodes is identified if a regulation relationship exists between the two corresponding genes. Such networks are called Gene Regulatory Networks (GRNs). Their construction as well as their analysis remain challenging despite the large number of available inference methods.
In this thesis, we propose to address this network inference problem with recently developed techniques pertaining to graph optimization. Given all the pairwise gene regulation information available, we propose to determine the presence of edges in the final GRN by adopting an energy optimization formulation integrating additional constraints. Either biological (information about gene interactions) or structural (information about node connectivity) a priori have been considered to restrict the space of possible solutions. Different priors lead to different properties of the global cost function, for which various optimization strategies, either discrete and continuous, can be applied. The post-processing network refinements we designed led to computational approaches named BRANE for "Biologically-Related A priori for Network Enhancement". For each of the proposed methods — BRANE Cut, BRANE Relax and BRANE Clust — our contributions are threefold: a priori-based formulation, design of the optimization strategy and validation (numerical and/or biological) on benchmark datasets from DREAM4 and DREAM5 challenges showing numerical improvement reaching 20%.
In a ramification of this thesis, we slide from graph inference to more generic data processing such as inverse problems. We notably invest in HOGMep, a Bayesian-based approach using a Variational Bayesian Approximation framework for its resolution. This approach allows to jointly perform reconstruction and clustering/segmentation tasks on multi-component data (for instance signals or images). Its performance in a color image deconvolution context demonstrates both quality of reconstruction and segmentation. A preliminary study in a medical data classification context linking genotype and phenotype yields promising results for forthcoming bioinformatics adaptations.
Resume
Dans de nombreuses applications telles que la médecine, l'environnement ou les biotechnologies, la découverte de nouveaux processus de régulation des gènes améliore la compréhension des réponses phénotypes à des stimuli extérieurs. Pour cela, il est d'usage de générer et d'analyser des données transcriptomiques obtenues via des puces à ADN ou plus récemment des expériences RNAseq. Ces données correspondent, pour tous les gènes d'un organisme placé dans différentes conditions expérimentales, à leurs niveaux d'expressions. À partir de ces données, les mécanismes de régulation des gènes peuvent être retrouvês via des liens topologiques dans des graphes. Dans de tels réseaux, les noeuds correspondent aux gènes. Un lien entre deux noeuds est identifié si une relation de régulation existe entre les deux gènes correspondant. Ces réseaux sont communément appelés Réseaux de Régulation de Gènes (RRGs). Malgré une profusion de méthodes d'inférence de RRGs, leur construction et analyse restent encore à ce jour un défi.
Dans ce travail de thèse, nous proposons de traiter le problème d'inférence de RRGs par des méthodes d'optimisation dans des graphes. Disposant d'un score d'interaction pour tous les couples de gènes, nous déterminons la présence d'arêtes dans le RRG final via la formulation d'une fonction objectif, intégrant des contraintes, à optimiser. Notamment, des a priori biologiques et/ou structuraux ont été considérés pour restreindre l'espace des solutions possibles. Les différents a priori émulent des propriétés différentes de la fonction objectif, pour laquelle diverses stratégies d'optimisation, discrète et/ou continue, peuvent être utilisées. Les stratégies de post-traitement que nous avons proposé pour l'amélioration d'inférence de réseaux a mené à un ensemble d'approches nommé BRANE pour "Biologically-Related A priori for Network Enhancement". Pour chacune des méthodes proposées — BRANE Cut, BRANE Relax et BRANE Clust — nos contributions sont triples : formulation basée sur des a priori , adaptation de la stratégie d'optimisation et validation (numérique et biologique) montrant des améliorations atteignant 20% sur des données de parangonnages issues des challenges DREAM4 et DREAM5.
Dans une vision complémentaire, nous avons également considéré des problèmes de traitements de données plus génériques, tel que les problèmes inverses. Nous avons notamment étudié HOGMep, une méthode Bayesienne résolu par des méthodes variationnelles. Cette approche permet de faire conjointement de la restauration et de la classification/segmentation sur des données multi-composantes (signaux ou images, par exemple). Les performances d'HOGMep, dans un contexte de déconvolution d'images couleurs, ont aussi bien montré des qualités de reconstruction que de segmentation. Une étude préliminaire pour de la classification de données médicales (dans un contexte liant phénotype et génotype) a donné des résultats prometteurs, intuitant des adaptations à venir en bioinformatique.